IRC
This method keyword requests that a reaction path be followed. The initial geometry (given in the molecule specification section) is that of the transition state, and the path can be followed in one or both directions from that point. The forward direction is defined as the direction the transition vector is pointing when the largest component of the phase is positive.The geometry is optimized at each point along the reaction path. (...)
IRC calculations require initial force constants to proceed. You must provide these to the calculation in some way. The usual method is to save the checkpoint file from the previous frequency calculation (used to verify that the optimized geometry to be used in the IRC calculation is in fact a transition state), and then specifiy IRC=RCFC in the route section. (...)
IRC calculations accept Z-matrices and Cartesian coordinates as molecule specifications.
Path Selection Options
Forward: follow the path only in the forward direction, where "forward is defined as the direction the transition vector is pointing when the largest component of the phase is positive.Reverse: follow the path only in the reverse direction.
MaxPoints=N: number of points along the reaction path to examine (in each direction if both are being considered)
StepSize=N: step size along the reaction path, in units of 0.01 a.u.
MaxCyc=N: sets the maximum number of steps in each geometry optimization. The default is 20.
Coordinate System Selection Options
MassWeighted: follow the path in mass-weighted internal coordinates. MW is a synonym for MassWeighted. This is the defaultInternal: follow the path in internal (Z-matrix) coordinates. Cartesian: follow the path in Cartesian coordinates.
Needless to say, computations of reaction pathways are multi-part processes. You have the option of doing each part of the computation as a single job, or you can run all parts together as one job using some the Link options that were described in the geometry optimization reading. Fundamentally, reaction pathway analysis is a three step process:
The first step is to run an optimization of the transition state. One possible transition state might be the hydrogen elimination to give H2 and CO. Hehre ("Ab Initio Molecular Orbital Theory") describes the reaction as follows:
The elimination reaction
H2CO ---> H2 + CO
has also been investigated. Least-motion removal of H2 from formaldehyde,
while retaining C2v symmetry is forbidden by orbital symmetry
considerations; the true transition structure (below) distorts to
Cs symmetry:
C----O
/ \
/ \
H-----H
C2----O1
/ \
/ X3
/
H4-----H5
%Chk=H2CO
# RHF/6-31G Opt(CalcFC,TS) Test
H2 + CO <-> H2CO (Opt)
0,1
O
C,1,R2
X,2,1.,1,A3
H,2,R3,3,A3,1,180.
H,4,R4,2,A4,1,0.
Variables:
R2=1.20
R3=1.13
R4=1.00
A3=78.
A4=85.
Even though we are fairly certain that this is a valid transition state, it is important to characterize the stationary point found in the optimization of the transition geometry. It is here that a frequency calculation is used. We can save computing time by using the force constant values and the optimized geometry calculated by the optimization run through the use of the Guess=Read and Geom=Checkpoint keywords. As mentioned in the previous section, it is important to always use the same basis set throughout the run.
%Chk=H2CO # RHF/6-31G FREQ Guess=Read Geom=Checkpoint Test H2 + CO <-> H2CO (Freq) 0 1Remember that you still must include the charge and multiplicity, followed by a blank line. The molecular geometry specification is being retrieved from the checkpoint file.
The output for this section shows one imaginary frequency:
****** 1 imaginary frequencies (negative signs) ******
Harmonic frequencies (cm**-1), IR intensities (KM/Mole),
Raman scattering activities (A**4/AMU), Raman depolarization ratios,
reduced masses (AMU), force constants (mDyne/A) and normal coordinates:
1 2 3
A' A' A"
Frequencies -- -2185.2500 781.7490 1044.2087
Remember that you can look in the job archive at the end of the run, to see if NIMAG is set to 1, which is the number of imaginary frequencies:
Test job not archived. 1\1\NCSC-FLYER\Freq\RHF\6-31G\C1H2O1\TNG60\17-Aug-1995\0\\# RHF/6-31G FREQ GUESS=READ GEOM=CHECKPOINT TEST\\H2 + CO <-> H2CO (Freq)\\0,1\O,- ...stuff deleted ]\NImag=1\\0.03513009,0.,0.01984599,0.04205894,0.,1.09716234,0.0036780At this point you are ready to run the IRC calculation. Again, we can reduce computational time by getting many of the input values from the checkpoint file. The RCFC option, if you recall, gets the Cartesian coordinates from the frequency calculation.
%Chk=H2CO # RHF/STO-3G IRC=RCFC Guess=Read Geom=Checkpoint Test H2 + CO <-> H2CO (IRC) 0 1The output from this run looks very similar to that of an optimization run:
IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC-IRC
------------------------------------------------------------------------
Z-MATRIX (ANGSTROMS AND DEGREES)
CD Cent Atom N1 Length/X N2 Alpha/Y N3
Beta/Z J ------------------------------------------------------------------------
1 1 O 0 -0.134614 0.000000 -0.682568
2 2 C 0 -0.134713 0.000000 0.461888
Here you see the line "IRC-IRC-IRC", followed by the various input geometries and calculations. IRC calculations then provide you with a table of results:
----------------------------------------------------------------------
SUMMARY OF REACTION PATH FOLLOWING:
(Int. Coord: Angstroms, and Degrees)
----------------------------------------------------------------------
ENERGY RX.COORD X1 Y1 Z1
1 -113.65874 -0.59922 -0.14757 0.00000 -0.68561
2 -113.65061 -0.49923 -0.14623 0.00000 -0.68535
3 -113.64358 -0.39924 -0.14493 0.00000 -0.68507
4 -113.63783 -0.29926 -0.14366 0.00000 -0.68477
5 -113.63354 -0.19927 -0.14243 0.00000 -0.68447
6 -113.63085 -0.09928 -0.14125 0.00000 -0.68417
7 -113.62988 0.00000 -0.14008 0.00000 -0.68387
8 -113.63087 0.09940 -0.13898 0.00000 -0.68356
9 -113.63369 0.19938 -0.13798 0.00000 -0.68329
10 -113.63833 0.29937 -0.13705 0.00000 -0.68305
11 -113.64471 0.39936 -0.13618 0.00000 -0.68285
12 -113.65268 0.49935 -0.13537 0.00000 -0.68269
13 -113.66199 0.59934 -0.13461 0.00000 -0.68257
X2 Y2 Z2 X3 Y3
1 -0.06787 0.00000 0.48192 -0.00978 0.00000
2 -0.07434 0.00000 0.48038 0.03114 0.00000
3 -0.08067 0.00000 0.47884 0.07274 0.00000
4 -0.08685 0.00000 0.47731 0.11497 0.00000
5 -0.09288 0.00000 0.47576 0.15780 0.00000
6 -0.09872 0.00000 0.47418 0.20124 0.00000
7 -0.10462 0.00000 0.47250 0.24701 0.00000
8 -0.11030 0.00000 0.47077 0.29332 0.00000
9 -0.11556 0.00000 0.46907 0.33826 0.00000
10 -0.12061 0.00000 0.46732 0.38362 0.00000
11 -0.12547 0.00000 0.46553 0.42930 0.00000
12 -0.13016 0.00000 0.46373 0.47520 0.00000
13 -0.13471 0.00000 0.46189 0.52126 0.00000
Z3 X4 Y4 Z4
1 1.53741 1.43952 0.00000 1.01408
2 1.53383 1.45431 0.00000 1.03186
3 1.52957 1.46734 0.00000 1.04991
4 1.52488 1.47859 0.00000 1.06822
5 1.52002 1.48802 0.00000 1.08678
6 1.51524 1.49548 0.00000 1.10554
7 1.51065 1.50135 0.00000 1.12525
8 1.50666 1.50514 0.00000 1.14907
9 1.50357 1.50696 0.00000 1.16412
10 1.50161 1.50694 0.00000 1.18313
11 1.50072 1.50535 0.00000 1.20202
12 1.50099 1.50246 0.00000 1.22074
13 1.50244 1.49866 0.00000 1.23927
----------------------------------------------------------------------
TOTAL NUMBER OF GRADIENT CALCULATIONS: 26
TOTAL NUMBER OF POINTS: 12
AVERAGE NUMBER OF GRADIENT CALCULATIONS: 2.16667
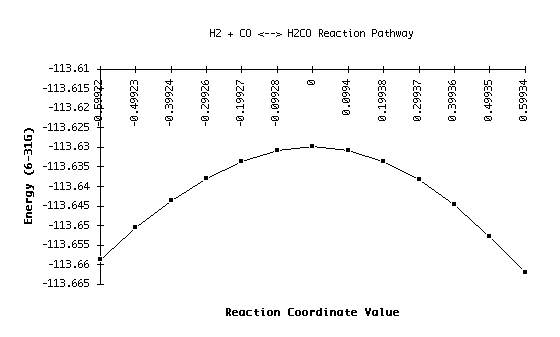
The items are shown in order of increasing distance along the reaction coordinate. For example, structure 1, with an energy of -113.65874, represents the dissociated side of the reaction. The transition state, or top of the barrier, can be identified where the reaction coordinate is equal to zero; in this data set, that puts the energy at -113.62988. The structure at item 13 represents the opposite side of the reaction coordinate curve. We can improve upon this calculation by changing the step size of the IRC run, and/or by increasing the number of steps calculated at runtime. Needless to say, both options increase computational time! The results of this run are shown on the graph below:
For this particular run, I have an activation energy of 0.02886 hartrees in one direction and 0.03211 hartrees in the other. These are relatively close, as the graph also suggests. Values closer to experimental could be reached with the use of better basis sets, but these are sufficient to give us a sense of the behavior of this particular reaction.
 The Shodor
Education Foundation, Inc.
The Shodor
Education Foundation, Inc.