Link0 section
The Link0 section has the purpose of establishing scratch files to be used in the computation. The most common scratch file is the "checkpoint" file. The checkpoint file serves to store useful intermediate datasets, such as structures, wavefunctions, and force constants. Generally, the checkpoint files are useful only to the program, not to you as output files to be evaluated. There are, however, several utility programs, such as "formchk", which can be used to format the checkpoint files for analysis by human eyes or visualization software. A checkpoint file can be requested by the use of this command in the Link0 sections:
%Chk=filename
We will probably not make use of this option, in which case, that section can be left out entirely.
This section controls the computation by selecting the type of approximation
and basis set to be used. The route section is specfied by the "#" character,
and is followed by a blank line. The route section consists of a wide
variety of keywords and options for controlling the computation.
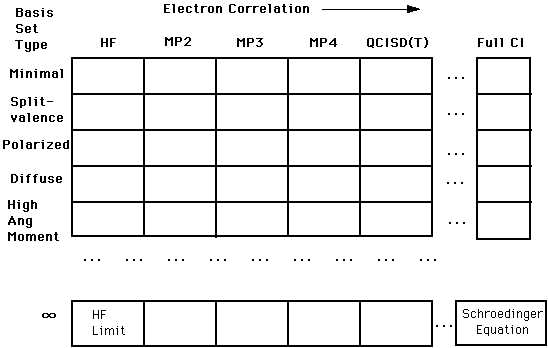
Approximation method: the approximation method is known as the "model
chemistry". For the simple problems in this session, we'll use the Hartree-Fock
approximation. You should recall that this method is the simplest and least
accurate method, in that it does not take into account electron-electron
correlations. It is, however, a good method for getting some insight into the
properties of a structure. There are two "versions" of the Hartree-Fock model
chemistry: restricted and unrestricted. The restricted method (RHF) is used
for those molecules that have only paired
electrons. Unrestricted Hartree-Fock (UHF) is to be used for those structures that have lone electrons in the structure. The Hartree-Fock model chemistry is the
theory that will be the most useful for our purposes. Other model
chemistries with a brief description are:
Configuration Interaction (CI): briefly, full CI methods
represent a mixing of all possible electronic conditions of the structure. CI represents a highly rigorous computational technique.
Möller-Plesset Perturbation Theory (MPx, where x is a version
number): builds
on the Hartree-Fock model chemistry by including some higher electronic
approximations.
Your choice of model chemistry is specified in the Route section, such as:
# RHF
Basis set: your choice of basis set is also specified in the
Route section, separated from the model chemistry by a "/". Basis sets represent a starting "guess" on the mathematical "shape" of the wavefunction. Remember that the basis set represents a "kick-start" to the mathematical engine in Gaussian94. The combination of model chemistry and basis set dictates the complexity and accuracy of the solutions
obtained. Keep in mind that the "goal" is to get closer and closer to a
solution of the Schroedinger equation:
The finalized route section might look like these:
# RHF / STO-3G
In the first example, we are asking for a restricted Hartree-Fock approximation using the basis set STO-3G (Slater-type orbitals, 3 gaussians). In the second example, we are requesting the Hartree-Fock approximation with the 6-31G basis set, and we want Gaussian94 to optimize the geometry. In the third example, we are using Hartree-Fock, with 6h 6-31G polarized basis set. We want it to do a direct self-consistent field calculation, with a frequency calculation being performed, with the initial guess coming from a checkpoint file, and the initial geometry also coming from the checkpoint file.
Charge: a positive or negative integer indicating the
total charge on the
molecule. For example, the charge on water is 0; the charge on ammonia
(NH3+)
is 1.
Spin multiplicity, or multiplicity for short, is the same as the multiplicity that MacSpartan uses in its calculations. For our purposes, we are only concerned with multiplicities of 1 or 2. Molecules with all electrons in pairs have multiplicity of 1, while molecules with a single unpaired electron have multiplicity 2.
Now comes the fun part! We must provide a starting geometry of the molecule of interest. You have three options on how you might do this:
These numbers are in units of Angstroms.
Other Cartesian coordinates will be provided if you want to do some exercises with G94. Ask a presenter for more help if you need other coordinates.
This method follows this algorithm:
1. Place the first atom at the origin
2. Place the second atom along the Z-axis using a bond length
3. Place the third atom in the XZ-plane using a valence angle
(angle between
three atoms) and a bond length.
4. Place the fourth and subsequent atoms by defining a dihedral
angle with
the plane of three other atoms, a valence angle and a bond length. A
dihedral
angle is the angle between four atoms. That is, the dihedral angle for atoms ABCD is the angle between the plane containing atoms ABC and the plane containing atoms BCD.
So, each line follows the format
Atomname atom1 bondlength atom2 angle atom3 dihedral
However, not all of these specifications may be needed for the first two or three atoms. Atomname is usually the element symbol followed by an integer, as you will see below. "atom1", etc, are the atom names of other atoms to which the geometry details refer.
Below is a Z-matrix for hydrogen perioxide.
In this Z-matrix, the leftmost oxygen is designated as O1 (the numbers are
optional, but helpful). The second atom is oxygen O2, and has a bond length of
1.404 Angstroms from the first oxygen. The leftmost hydrogen is designated as H1;
its relationship to O1 is with a bond length of 0.942 angstroms, with an
H1-O1-O2 angle of 92.5 degrees. The second hydrogen, H2, has a bond
length of 0.942 angstroms from O2, has a valence angle of 92.5 degrees for
H2-O2-O1, and a dihedral angle (going counterclockwise) of -180 degrees for H2-O2-O1-H1.
0 1
In this Z-matrix, the variable "r" represents the bond length, and the
variable "a" represents the bond angle. One of the values of using a variables section is that it allows you to easily modify common values. It will also be used in performing geometry optimizations in later sessions.
Route section
# CIS
# MP2
# HF / 6-13G OPT
# HF / 6-31G* SCF=DIRECT FREQ GUESS=READ GEOM=CHECKTitle section
The title section is just a label for
you to use in keeping track of your output. The title is placed on a single line by itself, without any special characters preceeding it.Molecule specification
This section is used to describe the basic beginning
characteristics of the molecule to be studied: charge, spin multiplicity,
units, and coordinates of the molecule.
ATOM X Y Z
C(1) -0.227 0.204 0.079
O(2) 0.642 -0.584 -0.208
H(3) -1.303 -0.062 -0.017
H(4) 0.019 1.227 0.442
O1
O2 O1 1.404
H1 O1 0.942 O2 92.50
H2 O2 0.942 O1 92.500 H1 -180.00
Variables section
In this section, you can establish
variables that replace
numerical values in the Z-matrix. An example molecule specification for
water
is as follows:
O
H 1 r
H 1 r 2 a
Variables:
r 1.0
a 105.0
 The Shodor
Education Foundation, Inc.
The Shodor
Education Foundation, Inc.
Copyright © 1998
Last Modified:
Questions or comments about this page should be directed to gotwals@shodor.org